

Abstract
There is no standard therapy for patients with myelodysplastic syndromes (MDS) refractory to hypomethylating agents (HMA) and the median overall survival (OS) of HMA-refractory MDS patients with IPSS Intermediate-2/High-risk MDS is ~6 months. Here, we present data from a phase 2 clinical trial of selinexor, an oral, first-in-class Selective Inhibitor of Nuclear Export (SINE) compound that inhibits XPO1, in patients with MDS refractory to HMAs. XPO1 is the major nuclear export protein responsible for shuttling many key cellular regulators out of the nucleus and is overexpressed in many cancers. Preclinical studies of selinexor have shown that inhibition of XPO1 causes nuclear retention of wildtype p53 and disruption of the NF-κB pathway, both relevant targets for MDS. Moreover, selinexor has shown promising responses in acute myeloid leukemia, multiple myeloma, and Non-Hodgkin lymphoma. We therefore set out to determine the safety and efficacy of selinexor in MDS, and to study potential molecular correlates of response to selinexor.
Patients with MDS or MDS/MPN who were refractory to azacitidine or decitabine were included (defined as either progression after initial response or if no response after >6 cycles). The primary endpoint was overall response rate (ORR = CR + mCR + PR + HI) and secondary endpoints included safety, response duration, and OS. The starting dose was 35 mg/m2 PO twice-weekly in 28-day cycles. Bone marrow biopsies, bone marrow DNA and peripheral blood RNA were collected from serial samples and used for correlative analyses including next-generation targeted gene sequencing, RNA-seq, immunohistochemistry, and proteomics. A total of 25 patients were evaluable for safety and 19 were evaluable for response. Six patients were not evaluable for response based on unrelated death from concurrent illness (n=5) or withdrawal of consent (n=1) before completion of one cycle. The median age was 77 (range, 50-85) and the majority of pts were High- or Very High-risk by IPSS-R (72%) with >10% bone marrow blasts (70%).
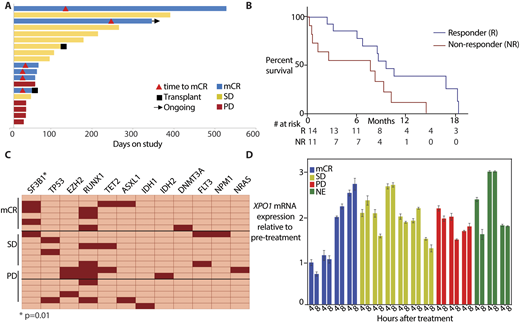
The ORR was 32% (all mCRs) with an average response duration of 6.8 months. An additional 42% of pts achieved stable disease (SD) for an average response duration of 6.4 months for all patients receiving clinical benefit (Figure A). The median overall survival was 9.7 months with a median follow-up of 2.1 years. Those with mCR or SD had greater survival than non-responders (Figure B). Grade 3 or 4 adverse events (AEs) in the first 3 pts (thrombocytopenia (n=3) and hyponatremia (n=1)) led to reduction of the dose to 60mg flat dose twice-weekly for 2 weeks followed by 1 week off, resulting in significantly improved tolerability. The most frequently occurring AE thereafter was grade 2 or 3 fatigue. All other AEs were manageable with routine supportive care.
The presence of an SF3B1 hotspot mutation was significantly associated with response to selinexor (p=0.01; Figure C). No other single mutation had a significant association, although patients with mutated TP53 and those with >3 mutations were numerically higher in the non-responders but did not reach statistical significance. The mRNA levels of a number of transcripts from the NF-κB pathway and of Myc were decreased after treatment with selinexor in responders, albeit with some variability. The levels of XPO1 mRNA increased with treatment within 4 hours of the fist dose, indicative of XPO1 target engagement with selinexor (Figure D). Further analyses are underway to determine if splicing is differentially modulated in SF3B1 mutated patients. Immunohistochemistry and proteomics for protein alterations are also ongoing.
These data indicate that selinexor is safe and tolerable in MDS when given at 60mg twice-weekly for 2 weeks with 1 week off with a 32% response rate. Responses appear to be enriched in patients with SF3B1 mutations and selinexor appears to affect pathways that may be activated downstream of mutant-SF3B1, such as NF-κB and Myc. Detailed correlative and pre-clinical analyses are ongoing and should provide further illumination of the biological relevance of these markers, which will be tested prospectively in future clinical trials. Further studies of selinexor combined with targeted agents that disrupt similar or complementary oncogenic pathways in MDS could further improve responses.
Rampal:Incyte: Honoraria, Research Funding; Stemline: Research Funding; Jazz: Consultancy, Honoraria; Constellation: Research Funding; Celgene: Honoraria. Park:Adaptive Biotechnologies: Consultancy; Kite Pharma: Consultancy; Amgen: Consultancy, Membership on an entity's Board of Directors or advisory committees; Pfizer: Consultancy; Shire: Consultancy; AstraZeneca: Consultancy; Novartis: Consultancy; Juno Therapeutics: Consultancy, Research Funding. Stein:Agios: Consultancy; Bayer: Consultancy; Novartis: Consultancy; Celgene: Consultancy; Daiichi Sankyo: Consultancy; Pfizer: Consultancy. Tallman:AbbVie: Research Funding; Cellerant: Research Funding; ADC Therapeutics: Research Funding; Daiichi-Sankyo: Other: Advisory board; Orsenix: Other: Advisory board; BioSight: Other: Advisory board; AROG: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal